This web site, https://www.vcru.wisc.edu/sdata, contains data from the laboratory of Philipp W. Simon, USDA-ARS Vegetable Crops Research Unit [Click here for our web page]
|
USDA ARS VCRU Data Server This web site, https://www.vcru.wisc.edu/sdata, contains data from the laboratory of Philipp W. Simon, USDA-ARS Vegetable Crops Research Unit [Click here for our web page] |
Programs in the bb project are now stored on GitHub at https://github.com/dsenalik/bb
Reference. If you use this software, you may cite using this reference:
Massimo Iorizzo, Douglas Senalik, Marek Szklarczyk, Dariusz Grzebelus, David Spooner and Philipp Simon
De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA
provides first evidence of DNA transfer into an angiosperm plastid genome
BMC Plant Biology 2012, 12:61
Download. Download bb.454contignet here - current version 1.0.7, May 4, 2012
Overview. This is a Perl program that will take an assembly of Roche 454 sequences generated by the Roche newbler/gsAssembler, and use the connection information to link generated contigs into a graphical map.
Description. A large amount of information about connections between various contigs in the gsAssembler assembly is contained in the 454ContigGraph.txt file generated by gsAssembler. I suggest looking at Lex Nederbragt's excellent description of the 454ContigGraph.txt file for more information about this file.
Thanks to Simon Gladman for adapting bb.454contignet to handle paired-end runs.
Use the parameters
A prerequisite for this program is the availablility of the
This is probably already installed on a standard Linux installation, but if not, the graphviz web size is
http://www.graphviz.org/
On Fedora you would just type
or on Ubuntu
Important. An important point during the gsAssembler assembly is to save
all contigs, no matter how small. Sometimes a very small,
even as small as 1 b.p contig can be found connecting two larger contigs, so discarding small contigs
could generate unnecessary gaps. Or, small indels between alleles could manifest themselves as very
small contigs. So, when creating your assembly in gsAssembler, make sure to set the
minimum contig size to
Example. Here is an example of a de novo chloroplast and mitochondrial genome assembly
from a single region (half of a plate) of 454 whole-genome shotgun sequence:
This assembly and image were used, after some manual enhancements, in the publication
cited above.
Example data files and commands:
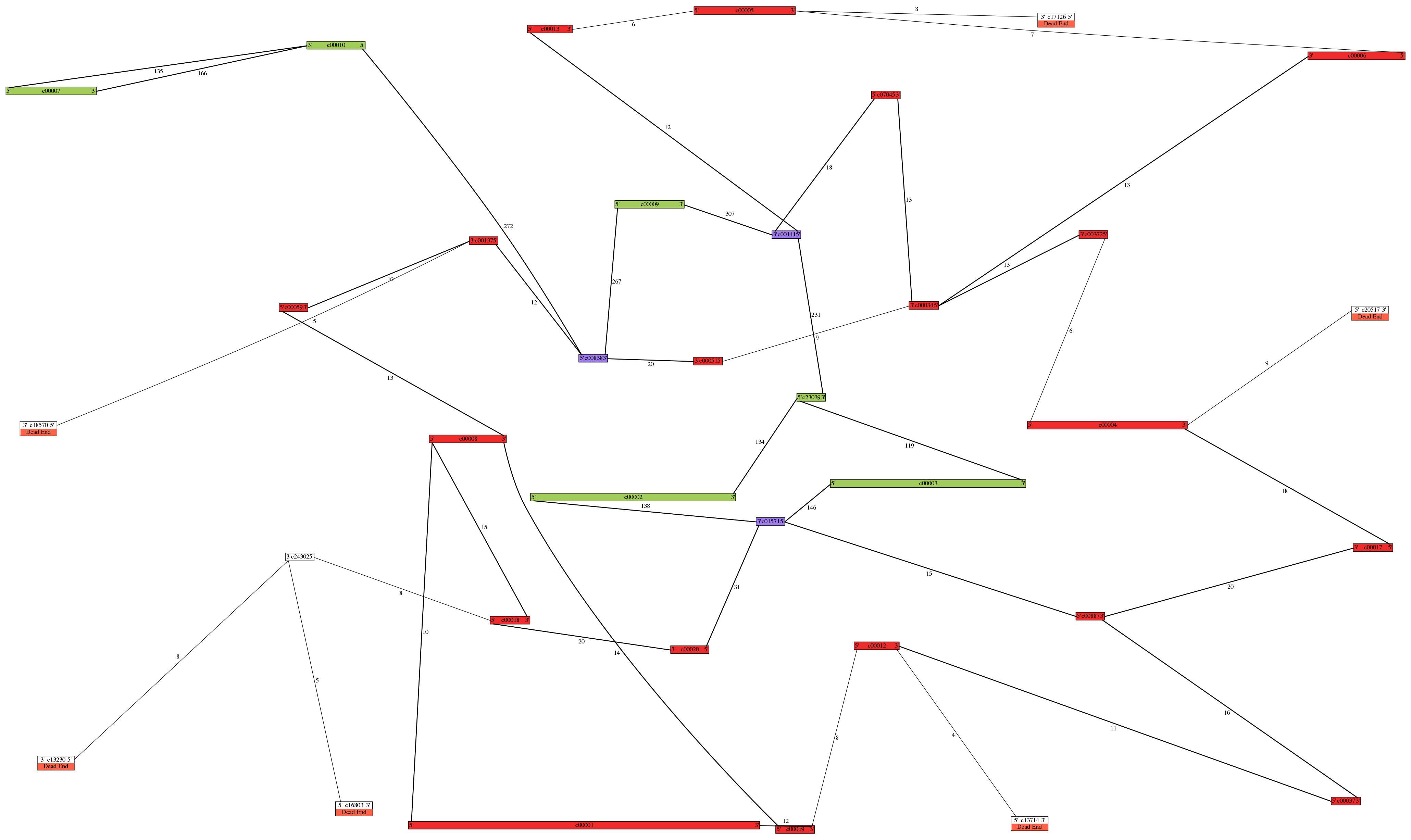
Color names are listed at http://www.graphviz.org/doc/info/colors.html
Here is the full program syntax, which you can obtain by typing the name of the program with no parameters:
bb.454contignet version 1.0.7
Required parameters:
--indir=xxx path to 454 assembly directory
--outfile=xxx output text file of results
--contig=xxx[,xxx]...
one or more starting contig numbers,
separated by comma, or multiple --contig
parameters may be used. Use just the
numeric portion of the contig
Optional parameters:
--type=xxx output file format, default is "png"
( anything besides "png" is experimental )
--cmdfile=xxx graphviz command file in .dot language will be created
using this name. If not specified, a temporary command
file will be created, and it will be deleted when done
--imgfile=xxx graph image file will be created with
this name. If not specified, will be
--outfile with .png extension added
--fastaout=xxx create a FASTA file of all contigs in
the output, save in this file
--abyssexplorer=xxx Generate a .dot file that can be used for
visualization with ABySS-Explorer 1.3.0,
http://www.bcgsc.ca/platform/bioinfo/software/abyss-explorer
--flowthrough include connection information derived from
reads that flow through more than two contigs
--flowbetween[=x] include connection information derived from
reads that flow from one contig into another
by default, if the distance value is zero, it will not be
shown, the optional value for this parameter is a minimum
distance, defaulting to 1, set to --flowbetween=0 to show
these links also
--pairlinks include connection information derived
from paired end reads, only applicable for assemblies
containing paired end reads
--alllinks sets --flowthrough, --flowbetween, and --pairlinks
--tag=tagname,contig[,contig]...
list of 1 or more contigs will be given
this tag. Multiple --tag allowed.
tagname is a text label that will be shown
in the final image, e.g. --tag="ATP1,14,34"
--label a synonym for --tag
--showbp show length in b.p. in graph
--shownt a synonym for --showbp
--showcoverage show average contig read coverage in graph
--color=colorname,contig[,contig]...
like --tag, but color the contig.
for list of valid color names see
http://www.graphviz.org/doc/info/colors.html
--forcelink=xxx-5:yyy-3 force a link where none exists
between specified ends, xxx and yyy are
contig numbers
--level=xxx maximum recursion level, default=2
--boldabove=xxx lines with read coverage >= this value
will be drawn in bold. no default value
--exclude=xxx[,xxx]...
one or contigs to never traverse past,
for example a repeated region contig
--listexcluded print out a list of which excluded contigs
are being ignored
--invert=xxx[,xxx]...
one or more contigs to plot backwards on
the graph, i.e. 3' to 5' direction
--extend=xxx auto extension for the single best
path, value is maximum steps, default=0
--lowlimit=xxx ignore connections < this number of reads
--highlimit=xxx ignore connections > this number of reads
--len=xxx len parameter to neato, default=1
--nolabel disable highlighting of dead ends, and limit
of recursion contigs
--overlapmode neato paramter, default is false, one of
none, true, scale
--nospline disable spline when edges would overlap
--help print this screen
--quiet only print error messages
--debug print extra debugging information
In place of lists of contigs, you can use @filename to read in
values for that parameter from a file, e.g. --exclude=@excl.txt
This program requires that the graphviz program "neato" be
available in the default PATH. The graphviz web site is
http://www.graphviz.org/
Version 1.0.7 adds experimental support for generation of a
Some other keywords for search engines: Roche 454 graph image, graph structure, edges, contig linkages, contig links, contig network, linked contigs, fork
Download bb.454contiginfo here - current version 1.0, March 21, 2012
This is a Perl program that will take an assembly of Roche 454 sequences generated by the Roche newbler/gsAssembler, and displays all information for one or more specified contigs, in particular, the connection and read flowthrough information.
Here is the full program syntax, which you can obtain by typing the name of the program with no parameters:
bb.454contiginfo version 1.0
This program analyzes some of the output files from a 454
assembly to find out everything available for a particular
contig. This information is all contained in the
454ContigGraph.txt file in the assembly directory.
Required parameters:
--infile=xxx input 454 assembly directory, or path
to 454ContigGraph.txt file
--contig=xxx contig to analyze ( multiple allowed )
use just the number e.g. --contig=123
or multiple numbers with , or ; as
separator, e.g. --contig=123,16389;599
--outfile=xxx output file name, use "-" for stdout
Optional parameters:
--showscaffold if contig is part of a scaffold, list
all contigs and gaps in that scaffold
--help print this screen
--quiet only print error messages
--debug print extra debugging information
Download bb.motif here - current version 1.0, June 14, 2010
This program was used to generate a supplemental file for the publication:
Marina Iovene, Pablo F. Cavagnaro, Douglas Senalik, C. Robin Buell, Jiming Jiang and Philipp W. Simon
Comparative FISH mapping of Daucus species (Apiaceae family)
Chromosome Research Volume 19, Number 4, 493-506, DOI: 10.1007/s10577-011-9202-y
A copy of the output file from the above publication: 10577_2011_9202_MOESM2_ESM.txt
This program will take one or more sequences in a FASTA
file, and look for your specified motif sequence in them.
Required parameters:
--motif=xxx nucleotide sequence of the motif to find
--infile=xxx name of input FASTA file, multiple allowed
--outfile=xxx name of summary file to create
Optional parameters:
--tbl2asnfile=xxx create a feature table for tbl2asn import
--tempdir=xxx save intermediate files in this directory.
If not specified, temporary files are not kept
--expect=xxx expect value for blast, default = 10.0
--debug debugging mode=extra info printed, keep temp files
--help print this screen
Download bb.orffinder here - current version 1.3.0 - Apr 1, 2013
This is a Perl program that will computationally detect
open reading frames in DNA or RNA sequences in FASTA format.
This is computationally similar to the NCBI program at
http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi,
but allows command-line automation of the process, as well as a few additional features.
This program will detect open reading frames in FASTA
DNA or RNA sequences. This is similar to the NCBI program at
http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi
Required parameters:
--infile=xxx input file name
--outfile=xxx output file name, use "-" for stdout
Optional parameters:
--fullstart use full set of start codons: ATG GTG CTG TTG
the default is to only use ATG
--anystart start of sequence is also a valid orf start
--minlen=xxx minimum orf length in b.p., default=100
--guessorientation guess orientation based on strand with the
most total orfs, this data will be be saved
instead of the list of orfs
--fasta output file is in FASTA format, each orf is
a separate sequence, information is in header
--nonorffasta second FASTA file with all sequence not in
the first one. File name is --outfile name
with "nonorf" inserted
--fastacollapse combine overlapping sequence in the FASTA file
--fastalargest if two orfs overlap, keep only the larger one
--trimheader remove any text in the FASTA header after
the first occurrence of white space
--origorder return list in sequence order instead of
the default which is sorted by size
--origorder=s same, but do + and - strands separately
--nsequence include a column with nucleotide sequence
--psequence include a column with protein sequence
--gffformat generate output in gff3 format. This also
enables --trimheader
--featureid=xxx column 3 of gff file, default is "CDS"
--non do not allow any "N"s in an orf
--help print this screen
--quiet only print error messages
--debug print extra debugging information
Download bb.fastareorder here - current version 1.0, September 3, 2011
This program will allow changing the order or orientation of multiple sequences in FASTA format, or extraction of a subset of sequences. The resulting sequences can optionally be concatenated into a single sequence.
bb.fastareorder Version 1.0
Rearrange the order of sequences in a FASTA file based on
your specified contigs and orientations
Required parameters:
--infile=xxx input FASTA file name with multiple sequences
--outfile=xxx output file name, or "-" for stdout
--seq=xxx sequences to keep, multiple allowed, a plus
"+" for forward orientation is optional,
or use "-" anywhere to indicate reverse
complement. Use ".." to indicate a range.
Use "," to separate entries. Examples:
--seq=contig45 --seq=46,49,-21..23
--seq=+32 --seq=-65 --seq=76-
--seq=00021..45- -seq=45+..47
or use "s" for a spacer of 20 Ns
e.g. --seq=00021+,S,45-
The --seq parameter may be omitted if --exclude
or --random is used instead
Optional parameters:
--exclude=xxx use this in place of the --seq parameter to
output all contigs except these. Order will be
unchanged from the original file in this case.
--random=xxx return this many sequences selected at random
and placed in random order
--coordinates create this output file, which will store
the starting and ending position of each contig
--onesequence concatenate all sequences into one
--blankline for --onesequence mode, put a blank line
between each sequence
--prefix=xxx if using --onesequence, use this prefix,
default = "concatenated"
--append append to existing --outfile
--startstop add starting and ending base position
to FASTA headers
--noqual if a .qual file is present, a corresponding
output .qual file is created. This flag
turns off this quality file processing
--help print this screen
--quiet only print error messages
--debug print extra debugging information
MITOFY was not created by us, but we provide a public web server
that can be used to run a MITOFY analysis.
This page can be accessed at
VCRU MITOFY Public Web Server
A download link may be found on that page.
The MITOFY home page is
http://dogma.ccbb.utexas.edu/mitofy/
This page last modified Monday, 11-Aug-2014 20:16:55 CDT